Математики придумали архиватор для ДНК
Математики из Массачусетского технологического института предложили новый способ хранения и обработки данных о последовательностях ДНК.
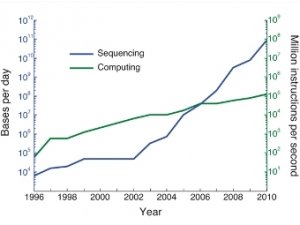
Он должен помочь справиться с наплывом данных от все большего числа
прочитанных геномов. Работа с описанием нового алгоритма опубликована в
журнале Nature Biotechnology, а ее краткое содержание можно прочитать на сайте института.
Алгоритм основан на том, что
последовательности ДНК между всеми организмами в той или
иной степени схожи, а наибольший интерес для ученых представляют
различия. Поэтому, по словам авторов, хранить и обрабатывать следует не
сами последовательности, а их отличия друг от друга.
Если, например, поиск определенной последовательности в геноме
некоторого организма уже проводился, то поиск той же последовательности в
новом геноме следует проводить не по всей последовательности, а только в
тех местах, где новый геном отличается от старого. Это позволяет существенно снизить время поиска последовательностей и нагрузку на вычислительные центры.
Разница в длительности вычислений между старым и новым алгоритмом
зависит от количества уже прочитанных геномов – чем их больше, чем
труднее искать по-старому и тем очевиднее преимущества нового алгоритма.
Упор на поиск различий в близких геномах соответствует современному
развитию биологии. С одной стороны, в последнее десятилетие ре
...
Читать дальше »